Stuck on You: Discovering Covalent Therapeutics with Biophysics
Covalent therapeutics are drug compounds that establish robust and often irreversible connections with their targets, allowing them to remain bound for extended periods compared to conventional non-covalent therapeutics.
This potentially has important advantages when developing new treatments. Key to this is the ability to produce an effect at a lower dose, which can be taken less often. Reduced dosing can decrease side effects and make new medicines more economical and easier to take.
Many drugs on the market have a covalent mode of action. Interestingly, many of these were initially found to function covalently long after their development and administration to patients, with Aspirin being a well-known example. But what makes the targeted discovery of new covalent therapeutics so challenging, despite there being many available already?
The challenges of covalent therapeutics
Whilst covalent therapeutics have many advantages, they also have an additional set of risks relative to non-covalent drugs. If a non-covalent drug binds to something other than its target, it will likely do so less well and dissociate before any effects are observed. Covalent molecules are different, once bound they tend to stay bound, meaning that non-target effects are more likely to be serious. It is the goal of any therapeutic pipeline, whether covalent or not, to maximise positive effects and minimise side effects. This can be more of a challenge with a covalent mode of action.
Filling in the gaps with biophysics
Covalent therapeutics are time-dependent. The formation of a covalent bond is relatively slow, which means that the apparent effectiveness of a molecule increases with more time. This is problematic for testing these molecules as this can drastically affect their activity.
- How long does it take to set up the assay?
- Can you guarantee that each step is performed for the same second each time?
- Can the temperature be the same every time too?
In practice, this is very difficult to do, but this is where SPR comes in. Surface Plasmon Resonance (SPR) is a biophysical technique that can observe molecular interactions in real-time. It is also tightly controlled in terms of temperature and contact time, both key parameters in determining the rate of covalent bond formation. This can be used to characterise compounds both from a selectivity perspective (the non-covalent part of an interaction) and reactivity (the covalent part of an interaction). By defining these parameters, we can better guide medicinal chemistry efforts to balance reactivity with selectivity and avoid issues later in the drug discovery process.
We use this platform in two ways, and it might come as a surprise that one of these approaches is to reduce the covalent part of the interaction so that it effectively disappears. This allows us to identify well-behaved compounds and remove any that have undesirable characteristics, which can sometimes be hidden by covalent activity. This is most important when looking for new hits and starting points for drug discovery, but its significance lessens as molecules become more potent and effective.
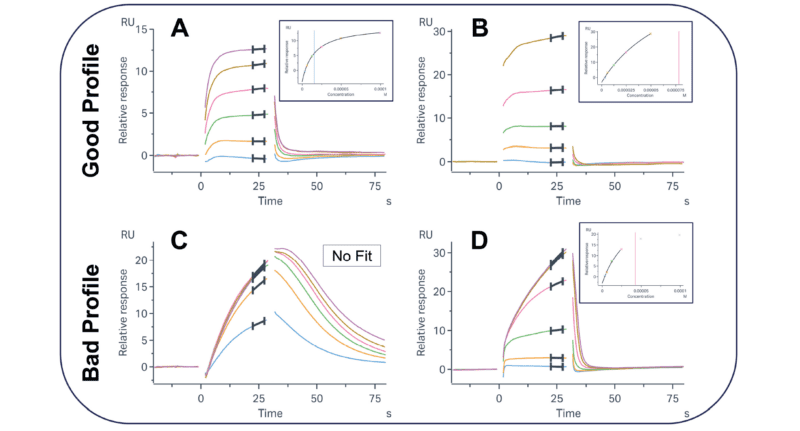
Figure 1: Example sensorgrams and binding profiles of desirable (A and B) and undesirable (C and D) binders to a target of interest. The selection of the most promising molecules for advancement relies on assessing both the overall sensorgram shape and the concentration-response curve. Molecules that are saturable and fully dissociating are preferred over those that are not at the hit-finding stage.
The second way of using this platform is to fully characterise a covalent binding interaction. This includes examining the initial non-covalent recognition of the binding site, followed by the formation of the covalent bond between the two molecules. This allows us to assess the balance of the molecule’s potency, how much is reactive vs. how much is target recognition and selectivity, providing valuable guidance for the effective development of covalent therapeutics.
The overall potency of a covalent molecule is defined by two parameters: kinact (the rate of covalent bond formation) and KI (the strength of the non-covalent interaction. We can determine the values of these using the “two-state” fitting model, common to many SPR analysis platforms. This model aligns directly with the well-established parameters that have been determined for covalent binding. By using SPR to directly observe these constants, we can provide full characterisation in a relatively high-throughput format compared to other techniques. This technique is universally applicable to all target classes and does not rely on any inherent activity of the target in question.
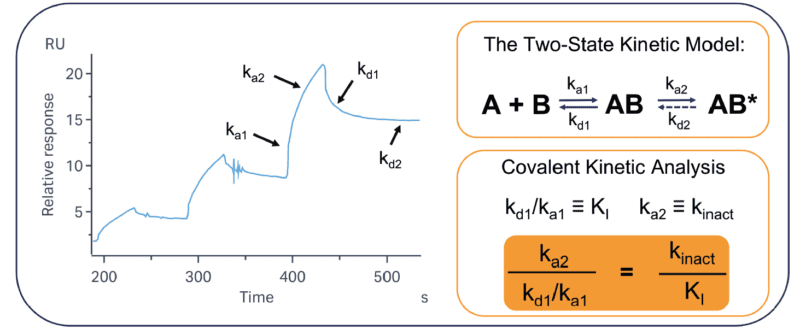
Figure 2: The two-state kinetic model aligns with the key parameters of covalent binding. Key to covalent interactions are the parameters kinact and KI. kinact is the rate of covalent bond formation and is primarily dictated by k2 (as k-2 will be negligible in most cases). KI is the non-covalent interaction and is quantified by k1 and k-1. The application of SPR data to the covalent inhibitor model allows for effective analysis of covalent binders. In most cases, it is assumed that kd2 = 0, but this can be adjusted for reversible covalent interactions. By using the two-state kinetic model for analysis, we can correlate the SPR kinetic output to the traditional covalent kinetic parameters.
However, this method comes with its challenges. The nature of covalent molecules means that they inactivate the captured protein surfaces, used in SPR, and potentially block other binders. To maintain throughput and efficiency, the use of regenerable and re-capturable surfaces is crucial and is something that we actively employ in our assays.

Download your free eBook: Successful In Vitro Strategies for Drug Discovery
Learn more about optimal cascade design, hit-to-lead phase insights and lead optimization strategies
Supporting Drug Discovery Pipelines
A targeted approach to covalent drug discovery often starts with a covalent fragment screen. These are primarily performed using in-tact Mass Spectrometry (MS) as the preferred approach, due to the high-throughput assessment of covalent bond formation as an addition of mass to the target molecule. These reactive fragments are very small and efficient molecules that can be precursors to drug-like molecules, although they can be more challenging to develop. Most drug discovery using fragments requires structural data to enable the development of the molecules with information on how they bind to their target. This can be more difficult with covalent molecules compared to non-covalent molecules, but it falls into the range of skills and technologies available at Sygnature.
Sygnature’s well-established biophysical platform for covalent drug discovery plays a crucial role, especially in the initial hit-finding stage, where validating high-quality hits prepares us for success in a drug discovery campaign. We can apply the platform throughout the life of a project, whether generating data for SAR activities, informing choices between therapeutic series or covalent warhead classes, or generating accurate characterisation data for therapeutic candidates. Our biophysical platform and expertise enable us to actively support covalent drug discovery pipelines with key data that can otherwise be difficult to produce.
About the author
Dr Philip Addis is a highly experienced biophysicist and leads the biophysics group at Sygnature Discovery. Philip has over 10 years of biophysics experience in biotech and CROs, and has supported drug discovery projects for small, medium, and large molecules across a wide range of therapeutic targets.